大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
近日,伦敦大学学院癌症研究所Nnennaya Kanu和弗朗西斯·克里克研究所Peter Van Loo团队合作在国际遗传学Top期刊《自然·遗传学》(Nature Genetics)发表题为“DNA methylation cooperates with genomic alterations during non-small cell lung cancer evolution“的重磅科研成果。该研究聚焦于非小细胞肺癌(NSCLC)进化过程中DNA甲基化与基因组变化的协同作用机制。团队通过整合分析多组学数据(RRBS、ChIP-seq、RNA-seq、WES等),揭示了DNA甲基化如何与基因组变化共同驱动NSCLC进化,并通过开发CAMDAC和MR/MN指标两个新的分析工具,提出变构染色质活性转变的新概念,为理解肿瘤进化提供了新视角。

标题:DNA methylation cooperates with genomic alterations during non-small cell lung cancer evolution(非小细胞肺癌进化过程中DNA甲基化与基因组改变的协同驱动机制)
发表时间:2025年9月10日
发表期刊:Nature Genetics
影响因子:IF29/Q1
技术平台:RRBS、ChIP-seq、RNA-seq、WES等(易基因金牌技术)
作者单位:伦敦大学学院癌症研究所、弗朗西斯·克里克研究所、美国安德森癌症中心、北京大学人民医院、伦敦玛丽女王大学等
DOI:10.1038/s41588-025-02307-x
在几乎所有癌症类型中,DNA甲基化都可能发生异常,但其在肿瘤进化过程中与基因组变化的互作尚未确定。为此,研究人员在前瞻性NSCLC多中心癌症TRACERx研究中对59名NSCLC患者的217个肿瘤区域和匹配的正常组织进行了简化基因组亚硫酸盐测序(Reduced Representation Bisulfite Sequencing, RRBS),以解析肿瘤甲基化。并开发了两个关键工具:CAMDAC(拷贝数感知甲基化去卷积分析)和MR/MN指标,用于DNA和RNA测序数据进行整合进化分析。其中,肿瘤内甲基化距离(Intratumoral Methylation Distance)量化了肿瘤内DNA甲基化的异质性。MR/MN指标根据调控性(MR)与非调控性(MN)CpG位点的高甲基化率对基因进行分类,以鉴定反复功能性高甲基化的驱动基因。研究结果揭示了与邻近原癌基因共扩增必需基因的DNA甲基化相关剂量补偿现象。研究人员提出了两种互补机制,这两种机制协同驱动受拷贝数改变影响的染色质经历类似变构活性转换的表观遗传变化,而处于正选择下的高甲基化驱动基因可能为患者的分层治疗提供了新的方法。
易小结
本研究填补了在肿瘤进化中DNA甲基化与基因组变化协同作用机制的研究空白。作者通过揭示DNA甲基化通过调控基因表达影响肿瘤进化,为开发新的治疗靶点和生物标志物提供了理论基础。此外,该研究还开创了基因组-表观基因组协同驱动研究的新范式,为未来类似研究提供了新的方法和思路。
易基因所提供的多组学产品(如本研究中的RRBS、ChIP-seq、WES和RNA-seq等)综合应用可以提供多维度的数据,为肿瘤研究提供全面的视角。如构建肿瘤发生和发展的综合模型,揭示基因组、表观遗传和转录调控的协同作用。不仅在肿瘤的早期诊断、精准治疗和预后评估中发挥重要作用,还为开发新的治疗策略和生物标志物提供更有力的支持,推动肿瘤研究和临床实践的进一步发展。
易基因相关产品拓展性案例展示
- 项目文章|Nature子刊:RRBS等揭示IDH1-R132H突变通过DNA甲基化加剧顺铂诱导肾毒性
- 项目文章 | ChIP-seq等揭示糖皮质激素和TET2共调控促进癌症转移的表观遗传机制
- 项目文章 | Nat Commun:中南大学曾朝阳/熊炜/龚朝建团队利用ChIP-seq等揭示头颈鳞癌免疫逃逸机制
- 项目文章 | Adv Sci:NSUN2介导m5C修饰代谢重编程促进肿瘤进展 揭示治疗新选择
- 项目文章|CDD:ChIP+RNA-seq技术揭示NURR1在前列腺癌从基因转录到肿瘤进展中的调控机制
- 项目文章|Hepatology/IF15.8:复旦中山医院沈英皓利用ChIP-seq及多组学解析肝癌仑伐替尼耐药机制(国人佳作)
研究方法
样本收集与处理:研究团队收集了59名NSCLC患者的217个肿瘤区域及匹配正常组织样本。
多组学技术应用:
- RRBS:解析DNA甲基化模式。
- ChIP-seq:研究染色质状态和转录因子结合位点。
- RNA-seq:分析基因表达水平。
- WES:检测基因组中的突变。
数据分析工具开发:
- CAMDAC:用于校正正常细胞污染对甲基化信号的影响。
- MR/MN指标:用于评估基因是否受DNA甲基化驱动的正选择。
无监督聚类分析:用于鉴定肿瘤样本的甲基化模式。
MethSig算法:用于鉴定甲基化驱动的癌症相关基因。
结果图形
(1)非小细胞肺癌(NSCLC)的肿瘤细胞特异性DNA甲基化图谱
通过对59名NSCLC患者的217个肿瘤区域及匹配正常组织的多区域简化基因组甲基化测序(RRBS),研究团队发现肿瘤样本的甲基化模式分为三类,分别对应正常组织、肺腺癌(LUAD)和肺鳞癌(LUSC)。启动子区的差异甲基化位点(DMPs)富集在分化相关基因(如SOX1、HOXD3)和抑癌基因(如SOX17、WT1-AS),提示甲基化可能通过沉默分化基因或抑癌基因促进肿瘤发生。此外,研究还发现肿瘤内甲基化距离(ITMD)指标,定量分析同一肿瘤不同区域的甲基化差异,结果显示肿瘤的ITMD值比正常组织高25倍,且与拷贝数变异异质性(SCNA-ITH)显著正相关,表明甲基化异质性与基因组不稳定性共同促进肿瘤克隆演化。这些发现揭示了非小细胞肺癌中肿瘤细胞特异性的DNA甲基化图谱,并强调其在肿瘤进化中的重要作用。
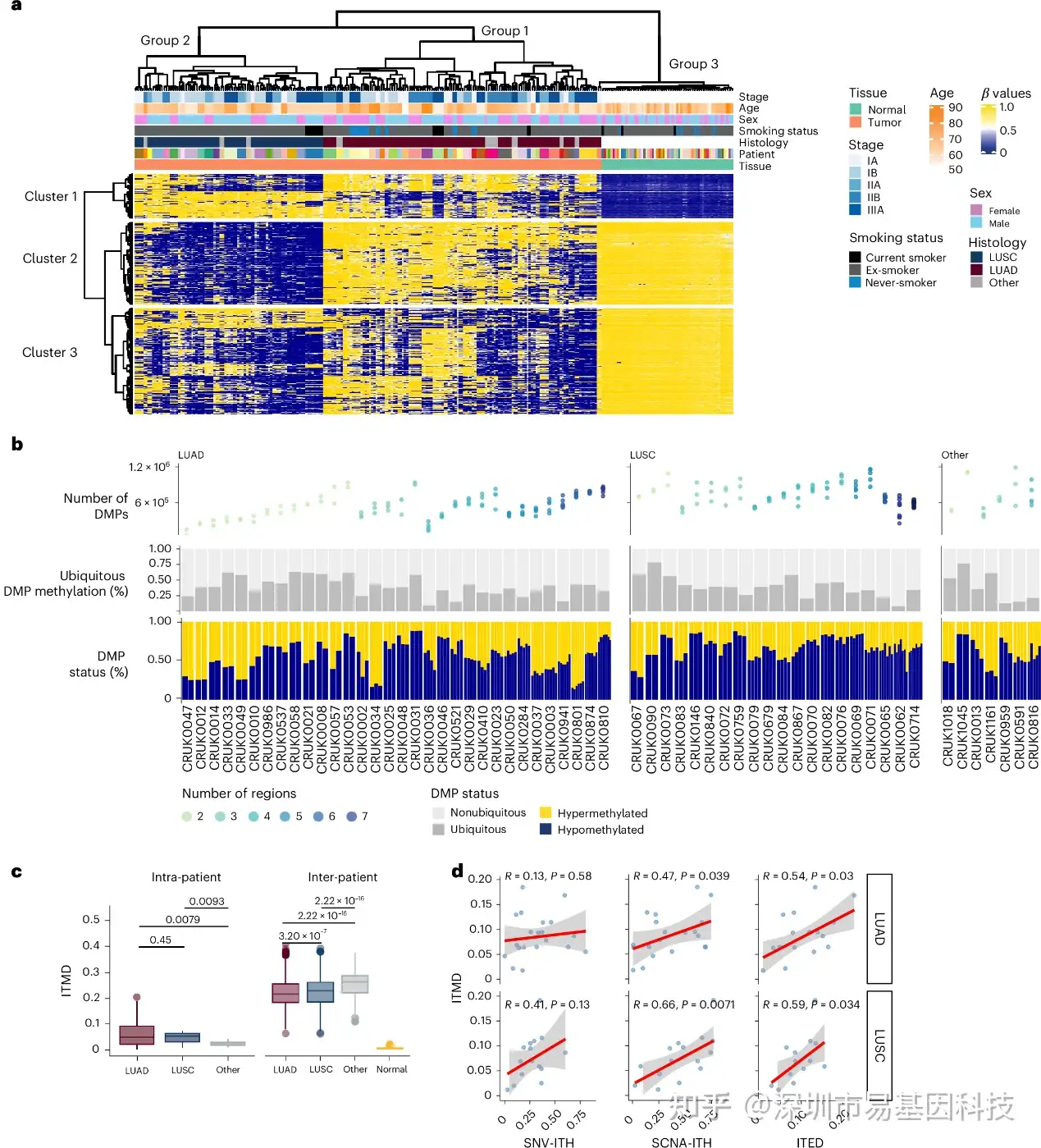
图1:TRACERx肺癌研究中的整体DNA甲基化图谱。
- 来自59名患者的217个肿瘤区域以及59个匹配的正常邻近组织(NAT)样本中5000个最易变异CpG位点的无监督聚类。黄色表示高甲基化CpG位点;蓝色表示低甲基化CpG位点。分组对应于患者样本,聚类对应于CpG位点。
- 展示了差异甲基化位点(DMPs)的数量、普遍DMPs(DMP存在的区域比例)的百分比以及DMPs的甲基化状态,表明了肿瘤内异质性(ITH)的程度。样本根据组织学亚型进行分层,并根据采样的区域数量从左到右按升序排列。
- 在肿瘤内(intra)和肿瘤间(inter)区域计算肿瘤内甲基化距离(ITMD)指标。
- ITMD评分与其他异质性指标的相关性;突变(SNV-ITH)、拷贝数变异异质性(SCNA-ITH)和转录本表达异质性(ITED),从左到右分别展示肺腺癌(LUAD,上)和肺鳞癌(LUSC,下)。拟合线表示通过稳健线性回归估计的平滑趋势,阴影区域表示95%置信区间。
(2)DNA甲基化对驱动基因表达的功能作用
研究团队通过分析发现,DNA甲基化对驱动基因表达有显著影响。在LUAD和LUSC中,约19%的抑癌基因(TSG)存在双重打击:同一肿瘤的不同区域同时发生拷贝数丢失和启动子高甲基化,二者协同抑制基因表达。此外,研究还发现当原癌基因(如KRAS、CCND1)因拷贝数扩增而激活时,邻近的必需基因(如RPS3、NOP2)会通过启动子高甲基化被剂量补偿,维持细胞内蛋白复合物稳态。这种现象被命名为“顺式变构染色质活性转换”(AllChAT),类似于生物化学中的变构效应,即局部基因组改变(拷贝数扩增)通过表观遗传修饰(甲基化)调控远端基因表达,确保肿瘤细胞活性。这些结果表明DNA甲基化不仅可以通过沉默抑癌基因促进肿瘤发生,还可以通过剂量补偿机制维持肿瘤细胞稳定。
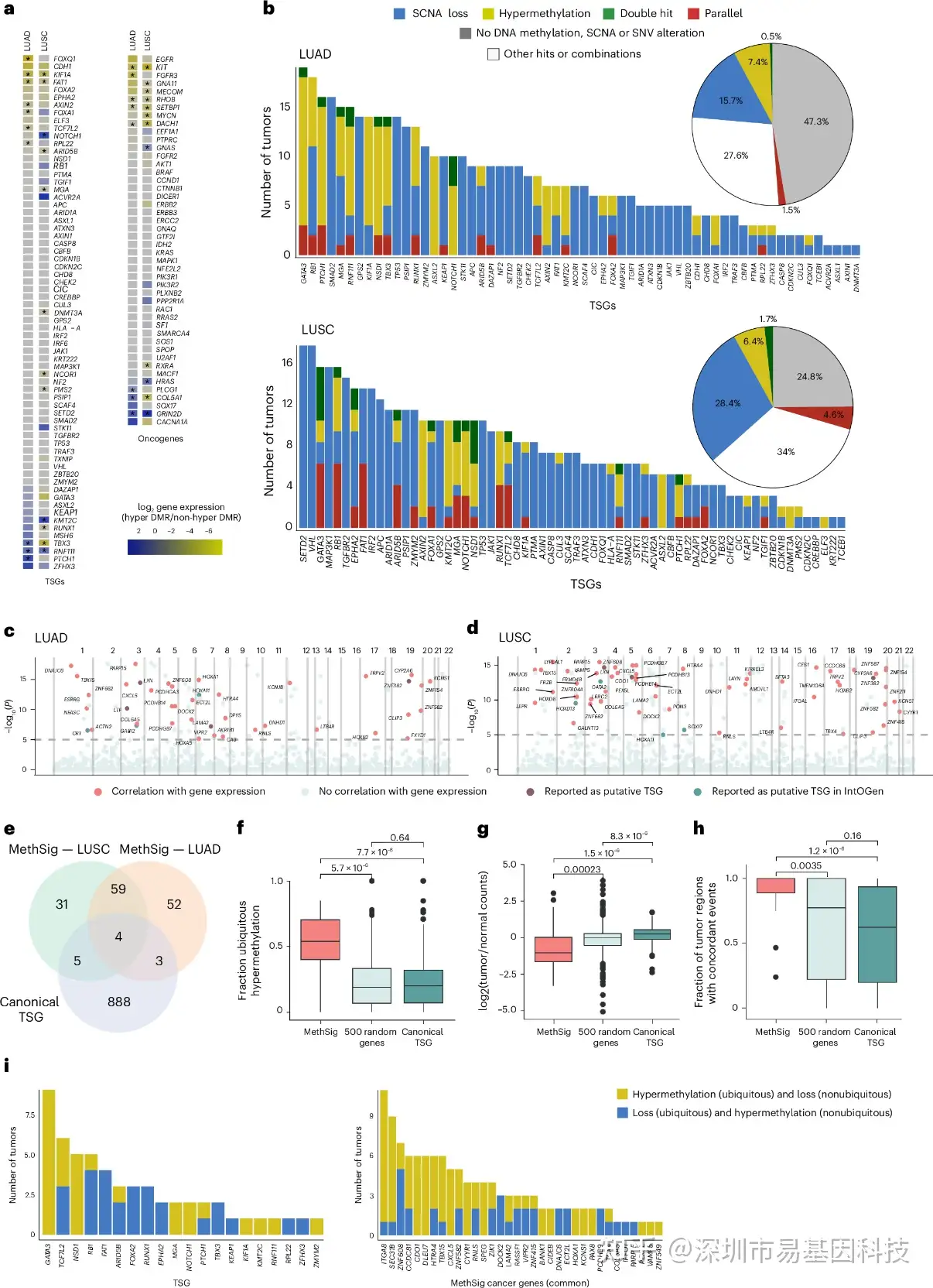
图2:DNA甲基化对驱动基因表达的影响分析。
- 分别靶向肺腺癌(LUAD)和肺鳞癌(LUSC)的基因组抑癌基因(TSGs,左侧)和原癌基因(右侧),启动子差异甲基化区域(DMR)状态对基因表达的影响。负值表示在基因启动子高甲基化(黄色)的样本中表达降低;正值表示在基因启动子高甲基化(蓝色)时表达增加。
- LUAD和LUSC肿瘤中基因组TSGs的拷贝数丢失(蓝色)或启动子高甲基化(黄色)数量。并行事件定义为在同一肿瘤的不同区域发生启动子高甲基化和拷贝数丢失(红色)。双重打击事件定义为在同一区域发生启动子高甲基化和拷贝数丢失的肿瘤(绿色)。其他事件组合包括拷贝数增加、突变或启动子低甲基化及其组合(白色)。饼图总结了所有基因组TSGs中每种事件类型比例。
c-d. 曼哈顿图展示LUAD(c)和LUSC肿瘤(d)中排名靠前的MethSig癌症基因。
e. 维恩图显示MethSig癌症基因与经典基因组TSGs的重叠。
f. 利用多区域DNA甲基化数据展示所有MethSig癌症基因、随机基因集和经典TSGs的普遍DNA高甲基化比例(t检验)。
g. MethSig癌症基因、随机基因集和经典TSGs在肿瘤与正常组织中的表达关系(t检验)。
h. MethSig癌症基因、随机基因集和经典TSGs中同时出现DNA高甲基化和拷贝数改变的区域占比。一致事件包括DNA高甲基化和拷贝数丢失,或低甲基化与拷贝数增加和扩增(t检验)。
- MethSig癌症基因和经典TSGs中具有普遍/非普遍DNA高甲基化和拷贝数丢失事件的肿瘤数量,用于确定这些改变在非小细胞肺癌中共现的相对时间顺序。
(3)DNA甲基化与拷贝数改变的差异
研究团队发现,DNA甲基化与拷贝数改变(CN alterations)之间存在显著差异。在某些情况下,DNA甲基化和拷贝数改变可以协同作用,如在抑癌基因的双重打击中,拷贝数丢失和启动子高甲基化共同抑制基因表达。但在其他情况下,这两种机制可能相互独立或存在复杂的相互作用。例如,在一些基因扩增区域,DNA甲基化可以通过剂量补偿机制调节邻近基因表达。这种差异表明DNA甲基化和拷贝数改变在肿瘤进化中既可以协同作用,也可以独立发挥作用,强调在研究肿瘤进化时需要综合考虑这两种机制。
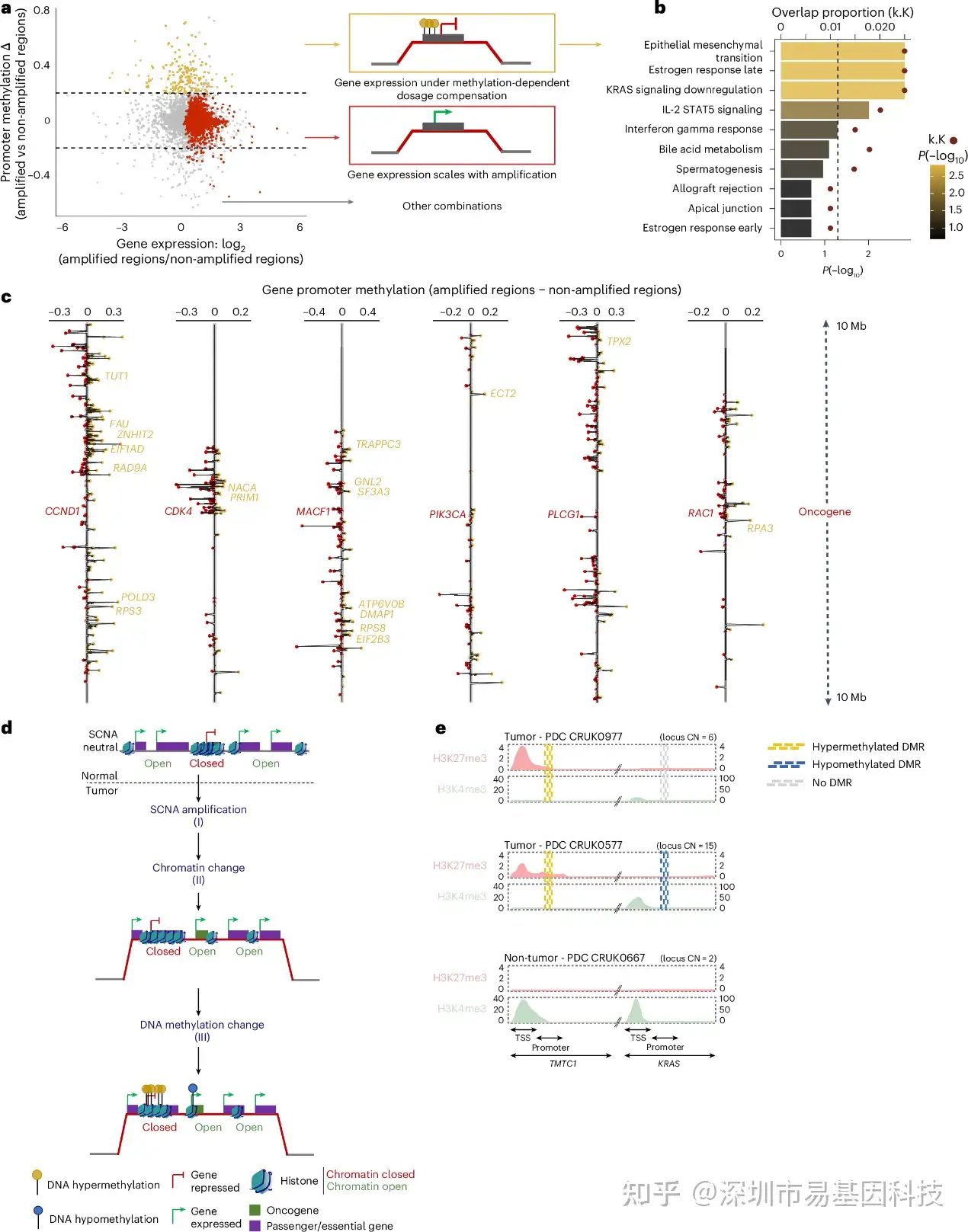
图3:DNA甲基化与拷贝数(CN)改变的差异性互作。
- 基因在扩增与未扩增时启动子甲基化中位数的差异(y轴)。大于0.2的值表示基因在扩增时DNA甲基化增加。x轴表示基因在扩增区域与未扩增区域之间表达量比率。正值表示基因表达与拷贝数扩增成正比。黄色突出显示基因可能处于DNA甲基化依赖的剂量补偿之下,因为其甲基化水平与拷贝数成正比。红色突出显示的基因的表达水平与拷贝数成正比,但与DNA甲基化不成正比。
- 潜在处于DNA甲基化依赖的剂量补偿之下的基因在癌症功能富集分析。
- 与拷贝数成正比的扩增原癌基因(红色)的表达水平,以及位于扩增原癌基因20 Mb范围内的样本之间基因启动子甲基化差异。从Achilles项目数据集中提取的必需基因用黄色标记。
- 示意图说明在原癌基因周围拷贝数改变与DNA甲基化之间的潜在协同作用。原癌基因位点的拷贝数变化可能触发局部AllChAT,影响共扩增必需基因和“乘客”基因(passenger genes)。
- 对基因对TMTC1作为扩增原癌基因KRAS的“乘客”基因进行AllChAT验证,在患者肿瘤来源原代细胞培养CRUK0977和CRUK0577中,以及来自患者CRUK0667的非肿瘤组织来源原代细胞培养中。位点拷贝数以数字形式表示。闭合染色质的抑制性组蛋白标记H3K27me3(红色)和开放染色质H3K4me3的活性组蛋白标记H3K4me3(绿色),并根据拷贝数对这两种组蛋白标记强度进行归一化。非肿瘤PDC作为对照,对每个基因启动子区域DNA甲基化状态进行评估。
(4)MR/MN指标筛选受DNA甲基化选择的基因
研究团队开发了MR/MN指标,类比基因进化中的“dN/dS”(非同义突变与同义突变比值),通过比较基因启动子区调控性CpG位点(甲基化后影响基因表达)与非调控性CpG位点(甲基化不影响表达)的甲基化率,评估基因是否受DNA甲基化驱动的正选择。基于MR/MN指标,研究团队进一步筛选出3个LUAD候选基因(CYP4F2、MSC、EIF5A2),其高甲基化状态与患者无病生存期(DFS)显著缩短相关(多因素Cox分析P<0.05)。这些基因的甲基化事件通常与经典抑癌基因(如STK11、CDKN1B)的突变共现,提示其可作为预测肿瘤进化轨迹的生物标志物。这一结果表明,MR/MN指标是一种有效的工具,可用于鉴定受DNA甲基化选择的基因,并为预测肿瘤进化和患者预后提供了新的生物标志物。
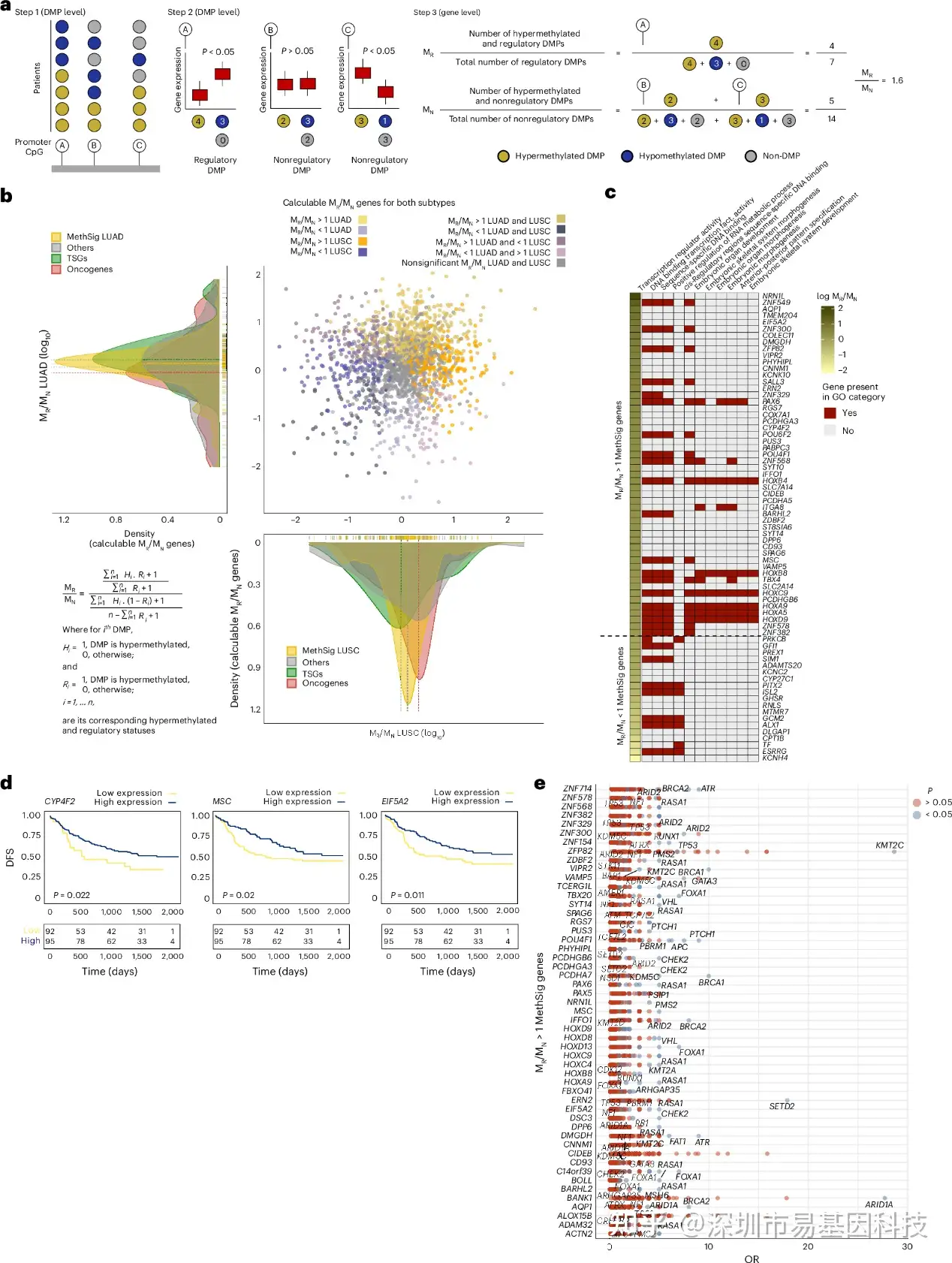
图4:通过将MR/MN应用于MethSig来识别癌症相关破坏事件。
- MR/MN指标开发示意图。(1)为队列中每个基因启动子中的CpG位点分配差异甲基化位点(DMP)状态。(2)根据CpG的高甲基化是否降低相应基因在队列中表达,将每个DMP表征化为调控性或非调控性。(3)根据每个基因启动子中调控性和非调控性CpG的聚合DNA甲基化状态,计算每个基因的MR和MN值。
- log–log散点图显示LUAD(y轴)和LUSC(x轴)中每个基因的常见可计算MR/MN比率。在密度图上,根据基因显示特定亚型的可计算MR/MN比率。
- 对MethSig基因进行GO功能富集分析,其中MR/MN> 1(上)和MR/MN< 1(下)。
- MR/MN>1的MethSig癌症基因(CYP4F2、MSC和EIF5A2)的表达的Kaplan-Meier曲线,与TRACERx队列中较短的无病生存期(DFS)相关。
- MR/MN>1的MethSig癌症基因的启动子DNA高甲基化事件与LUAD中经典抑癌基因驱动突变共现的比值比(OR)。
(5)受DNA甲基化破坏的癌症相关MethSig基因
通过MethSig算法(结合CAMDAC去卷积数据),研究团队鉴定出99个LUAD和118个LUSC的候选甲基化驱动基因(MethSig癌基因)。这些基因多富集于发育调控因子(如PAX6、TBX4)和HOX基因家族,且在肿瘤中呈现广泛甲基化(比经典TSG更普遍),提示其可能是肿瘤早期的表观遗传开关,通过沉默分化程序促进细胞恶性转化。这些MethSig基因的发现不仅为理解肿瘤早期进化提供了新的视角,还为开发新的治疗靶点和生物标志物提供了潜在的候选基因。
结论和启示
本研究通过整合RRBS、ChIP-seq、WES、RNA-seq等多组学数据,揭示了DNA甲基化与基因组变化在非小细胞肺癌进化中的协同作用机制,强调了表观遗传调控在肿瘤进化中的重要作用。研究结果不仅为理解肿瘤进化提供了新的视角,还为开发新的治疗靶点和生物标志物提供了理论基础。未来研究可以进一步探索其他表观遗传修饰与基因组变化的相互作用,为肺癌的精准治疗提供新的策略。此外,本研究开发的CAMDAC和MR/MN指标等分析工具,为未来类似研究提供了新的方法和思路,具有重要的应用价值。
关于易基因简化基因组甲基化测序(RRBS)研究解决方案
简化甲基化测序(Reduced Representation Bisulfite Sequencing,RRBS)是利用限制性内切酶对基因组进行酶切,富集启动子及CpG岛等重要的表观调控区域并进行重亚硫酸盐测序。该技术显著提高了高CpG区域的测序深度,在CpG岛、启动子区域和增强子元件区域可以获得高精度的分辨率,是一种准确、高效、经济的DNA甲基化研究方法,在大规模临床样本的研究中具有广泛的应用前景。
为适应科研技术的需要,易基因进一步开发了可在更大区域内捕获CpG位点的双酶切RRBS(dRRBS),可研究更广泛区域的甲基化,包括CGI shore等区域。
为助力适用低起始量DNA样本(5ng)量多维度甲基化分析,易基因开发了富集覆盖CpG岛、启动子、增强子、CTCF结合位点的甲基化靶向基因组测序方法:extended-representation bisulfite sequencing(XRBS),实现了高灵敏度和微量样本复用检测,使其具有高度可扩展性,并适用于有限的样本和单个细胞基因组CG位点覆盖高达15M以上。
技术优势:
- 起始量:100ng gDNA;
- 单碱基分辨率;
- 多样本的覆盖区域重复性可达到85%-95%、测序区域针对高CpG调控区域,数据利用率更高;
- 针对性强,成本较低;
- 基因组CG位点覆盖高达10-15M,显著优于850K芯片。
应用方向:
RRBS/dRRBS/XRBS广泛应用于动物,要求全基因组扫描(覆盖关键调控位点)的:
- 队列研究、疾病分子分型、临床样本的甲基化 Biomarker 筛选
- 复杂疾病及肿瘤发病机制等甲基化研究
- 模式动物发育和疾病甲基化研究
| 技术参数 | RRBS | DRRBS | cfDNA-RBS | Micro-RBS | sc-RBS |
|---|---|---|---|---|---|
| 原理 | MspI酶切+连接接头 | 多酶切+连接接头 | CCGG邻近片段连接接头 | CCGG邻近片段连接接头 | CCGG邻近片段连接接头 |
| 样本要求 | 1μg | 1μg | 1ng | 1ng | 单细胞/1-10个细胞 |
| 测序数据量 | 10G | 15G | 20G | 20G | 2G |
| 5XCG位点覆盖 | 6M | 8M | 6M | 10M | 4-8M |
关于易基因染色质免疫共沉淀测序 (ChIP-seq)
染色质免疫共沉淀(Chromatin Immunoprecipitation,ChIP),是研究体内蛋白质与DNA相互作用的经典方法。将ChIP与高通量测序技术相结合的ChIP-Seq技术,可在全基因组范围对特定蛋白的DNA结合位点进行高效而准确的筛选与鉴定,为研究的深入开展打下基础。
DNA与蛋白质的相互作用与基因的转录、染色质的空间构型和构象密切相关。运用组蛋白特定修饰的特异性抗体或DNA结合蛋白或转录因子特异性抗体富集与其结合的DNA片段,并进行纯化和文库构建,然后进行高通量测序,通过将获得的数据与参考基因组精确比对,研究人员可获得全基因组范围内某种修饰类型的特定组蛋白或转录因子与基因组DNA序列之间的关系,也可对多个样品进行差异比较。
应用方向:
ChIP 用来在空间上和时间上不同蛋白沿基因或基因组定位
- 转录因子和辅因子结合作用
- 复制因子和 DNA 修复蛋白
- 组蛋白修饰和变异组蛋白
技术优势:
- 物种范围广:细胞、动物组织、植物组织、细菌微生物多物种富集经验;
- 微量建库:只需5ng以上免疫沉淀后的DNA,即可展开测序分析;
- 方案灵活:根据不同的项目需求,选择不同的组蛋白修饰特异性抗体。
技术路线:
参考文献:
Gimeno-Valiente F, …, Van Loo P, Kanu N. DNA methylation cooperates with genomic alterations during non-small cell lung cancer evolution. Nat Genet. 2025 Sep 10. doi: 10.1038/s41588-025-02307-x.
相关阅读:
1.项目集锦 | 易基因近期染色质免疫共沉淀测序(ChIP-seq)研究成果
2.项目文章 | NAR:ChIP-seq等揭示蛋白质酰基化与c-di-GMP协同调控放线菌发育与抗生素合成机制
3. 项目文章|Plant Physiol:郑州果树所王力荣团队ChIP-seq等揭示桃树需冷量和芽休眠调控的关键基因
4.项目文章|Cell Rep:RRBS揭示MOF对UHRF1乙酰化修饰是DNA甲基化维持的关键调控机制
5.项目文章 | RRBS揭示基于DNA甲基化驱动基因的肾透明细胞癌预后模型的鉴定和验证
6.项目文章:泪腺RRBS+RNA-seq揭示Sjögren综合征相关干眼症的潜在基因
7.DNA甲基化修饰整体研究方案
8. DNA与蛋白质互作及染色质开放性研究方案
9.技术推介 | 简化基因组甲基化测序研究解决方案
10. 技术推介 | 染色质免疫共沉淀测序 (ChIP-seq)
机制研究专题
:价格、效果与分辨率)







+initializer_list 底层解析)
)
)


的核心板)





